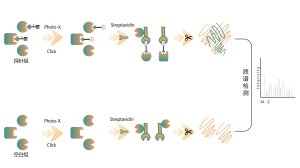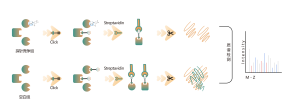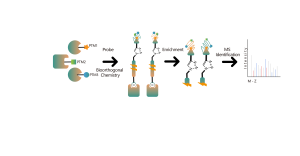
Produkte
Identifizierung von Proteinzielen durch differenzielle Proteomik
Technische Merkmale der Plattform
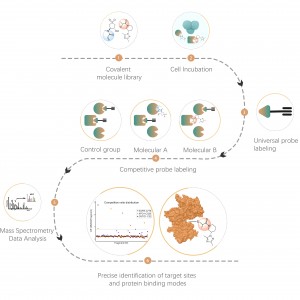
Die differenzielle Proteomik untersucht die Veränderungen des Proteoms unter verschiedenen physiologischen oder pathologischen Zuständen, wie z. B. Arzneimittelbehandlungen oder Genregulation, durch den Vergleich von zwei oder mehr Proben. Dieser Ansatz beleuchtet wichtige Lebensprozesse oder schwere Krankheiten, um die wichtigsten verschiedenen Proteine herauszufinden, die als Marker für qualitative und funktionelle Analysen gelten. Tausende von Proteinen können mit dem ChomiX-Standardprotokoll quantitativ identifiziert werden, einschließlich Proteomprobenvorbereitung, Proteaseverdau, Peptidfraktionierung, MS-Datenerfassung und Bioinformatikanalyse.

Serviceangebot
1. Quantitative und proteomweite Analyse unterschiedlicher Proteine, die durch Krankheiten, medikamentöse Behandlungen oder Umweltstress usw. verursacht werden.
2. Quantitative Analyse des Proteoms aus subzellulären Strukturen (Zellmembranen, Kerne, Mitochondrien usw.).
3. Entdeckung von Biomarkern im gesamten Proteommaßstab.
Proteomische Quantifizierungsmethoden
Markierungsfreie Quantifizierung (LFQ)
Technik:
Proteinquantifizierung durch Spektralzählung oder XIC-Intensität, Quantifizierung auf MS1-Ebene
Vorteile:
Keine Isotopenmarkierung, hoher Durchsatz
Probenanforderungen:
Zell-, Gewebe-, Blutproben etc.
Stabile Isotopenmarkierung durch reduktive Dimethylierung (ReDi)
Technologie:
Entweder reguläre (leichte) oder deuterierte (schwere) Formen von Formaldehyd und Natriumcyanoborhydrid werden verwendet, um zwei Methylgruppen an den N-Terminus des Peptids und die Seitenkette der Lysinreste hinzuzufügen. Quantifizierung auf MS1-Ebene
Vorteile:
chemische Duplex- und Triplex-Markierung, niedrige Kosten, schnelle Reaktionsgeschwindigkeit, hohe Reproduzierbarkeit, keine Probenbeschränkung;
Probenanforderungen:
Zell-, Gewebe-, Blutproben etc.
Stabile Isotopenmarkierung durch Aminosäuren in Zellkulturen (SILAC)
Technologie:
Zellkultur in Medium, das essentielle Aminosäuren mit stabiler Isotopenmarkierung zur Proteomquantifizierung in verschiedenen Proben enthält; Quantifizierung auf MS1-Ebene
Vorteile:
Duplex-Stoffwechselmarkierung, weniger Systemfehler;
Probenanforderungen:
lebende Zellproben.
Tandem-Massen-Tags (TMT/IBT)
Technologie:
Relative Quantifizierung der Peptidintensität durch ihre Reportergruppe in verschiedenen Proben; Quantifizierung auf MS2-Ebene
Vorteile:
Bis zu 16 Probenquantifizierung, genaue Quantifizierung;
Probenanforderungen:
Zell-, Gewebe-, Blutproben etc.
Abbildungslegende
Projektziel
Vergleichende Analyse der Veränderungen der gesamten Proteomspiegel zwischen der mit Arzneimitteln behandelten Gruppe und der Kontrollgruppe, um die molekularen Mechanismen zu untersuchen, die dem Arzneimittelphänotyp zugrunde liegen.
Beispieltypen
Zellproben, die Arzneimittel- und Kontrollbehandlungen unterzogen wurden und jeweils drei biologische Replikate umfassen.
Experimentelle Methode
Quantitative Identifizierung unterschiedlich exprimierter Proteine auf der Ebene des gesamten Proteoms mithilfe der TMT-basierten Proteomics-Methode zur Mehrfachisotopenmarkierung.
Datenvisualisierung

Vulkandiagramm zur Analyse der Proteinhäufigkeit
Wie im Vulkandiagramm gezeigt, wurden 5987 Proteine in insgesamt sechs Gruppen quantifiziert. Das Verhältnis jedes Proteins wurde mittels T-Test-Analyse analysiert und zeigte, dass in der mit Medikamenten behandelten Gruppe 560 Proteine hochreguliert und 363 Proteine herunterreguliert waren. Die zugehörigen Intensitätsinformationen wurden auch in der Heatmap dargestellt.

- Heatmap der Proteinhäufigkeit zwischen Kontroll- und Versuchsgruppe.
An den unterschiedlich exprimierten Proteinen, einschließlich GOTERM-Biological Process, GOTERM-Cellular Component und GOTERM-Molecular Function, wurden KEGG-Signalweg- und Genontologie-Analysen (GO) durchgeführt. Durch die Bewertung des Signifikanzniveaus der GOTERM-Anreicherung identifizierten wir funktionelle Kategorien und Signalwege, die durch die unterschiedlich exprimierten Proteine deutlich angereichert werden, und trugen so zur Erforschung der molekularen Mechanismen von Arzneimitteln bei.

- Genontologische Analyse unterschiedlicher Proteine.
Die Analyse der Genontologie, einschließlich GOTERM_ biologischer Prozess, GOTERM_ zelluläre Komponente, GOTERM_ molekulare Funktion und KEGG-Signalweg, ergab, dass die hochregulierten Proteine bei der Kernchromosomensegregation, der mitotischen Schwesterchromatidsegregation und dem Signalweg der Schwesterchromatidsegregation signifikant angereichert waren.