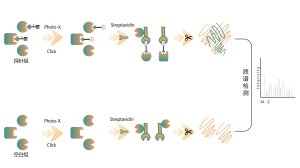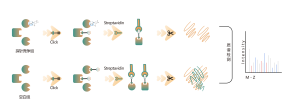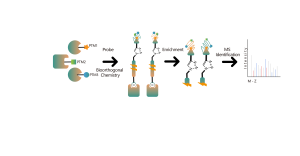
Productos
Identificación de regiones de interacción proteína-proteína, anticuerpo-antígeno: entrecruzamiento químico
Comprender la estructura y las interacciones de las proteínas es esencial para descubrir sus funciones biológicas. Debido a la complejidad del proteoma, ninguna técnica por sí sola puede revelar completamente estos aspectos. Los biólogos suelen utilizar una combinación de métodos, destacando la espectrometría de masas de reticulación (XL-MS) por sus ventajas únicas.
XL-MS proporciona información precisa sobre la distancia espacial entre residuos de aminoácidos y ofrece numerosos beneficios: requisito mínimo de muestra, bajas exigencias ambientales, sin limitaciones de peso molecular, reticulación in situ, facilidad de uso y amplia producción de datos.
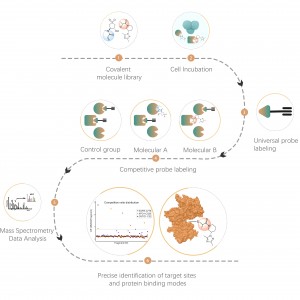
La tecnología XL-MS de ChomiX utiliza varios entrecruzadores químicos para apuntar a residuos de aminoácidos específicos, entrecruzando y analizando con precisión las interacciones proteína-proteína y anticuerpo-antígeno.
Plataforma Técnica
Los reticulantes químicos comunes (como se muestra en la figura siguiente) pueden entrecruzar covalentemente residuos de aminoácidos específicos cuando se agregan a sistemas de proteínas. Después de la digestión enzimática, se generan péptidos entrecruzados, que representan las regiones de interacción entre proteínas. Mediante la identificación por espectrometría de masas y el análisis preciso de las secuencias peptídicas, se puede obtener información sobre las regiones de interacción.

Reticulante de amino a tiol BS3

Reticulante aminosulfhidrilo EMCS

Nuestras ventajas
1. Excelencia profesional: nuestro equipo cuenta con una amplia experiencia y publicaciones en las principales revistas, ofreciendo servicios técnicos líderes en la industria.
2. Soluciones eficientes: Empleamos métodos confiables para impulsar los proyectos rápidamente, brindando soluciones sin preocupaciones.
3. Gestión de calidad rigurosa: al adherirnos a las normas ISO 9001, nuestro maduro sistema de gestión de calidad garantiza la autenticidad y confiabilidad de nuestros informes.
4. Gestión sistemática de proyectos: desde la consulta hasta la entrega de informes, proporcionamos actualizaciones oportunas del progreso, garantizando la satisfacción del cliente y la ejecución eficiente del proyecto.
5. Equipos de vanguardia: Equipados con espectrómetros de masas avanzados como Thermo Fisher Orbitrap Exploris 480 y Bruker timsTOF, facilitamos investigaciones innovadoras.
Nuestro Servicio
| Proyecto | Identificación de regiones de interacción para interacciones proteína-proteína y anticuerpo-antígeno. |
| Muestra | Proteína pura, complejo anticuerpo-antígeno. |
| Plataforma de hardware | Pulverizador de células ultrasónico sin contacto, sistema de imágenes ChemiDoc MP, espectrómetro de masas Orbitrap Fusion Lumos Tribrid/Orbitrap Exploris 480/Q Exactive HF-X/timsTOF Pro 2 |
| Duración del proyecto | 2-4 semanas |
| Entregables | Informe del proyecto (incluidos procedimientos experimentales, cuadros de análisis de datos, resultados de análisis bioinformáticos) |
| Precio | Haga clic para consultar |
Estudio de caso

En este caso, la proteína del suero BSA se entrecruzó usando el reticulante reactivo con amina BS3 para construir una estructura dimérica de la proteína del suero BSA. El análisis de espectrometría de masas de la estructura dimérica identificó segmentos de péptidos entrecruzados que coincidían con los datos de la literatura existente, validando así la precisión y confiabilidad del experimento.

Los péptidos entrecruzados realmente detectados son consistentes con los péptidos entrecruzados presentados en la siguiente literatura:Caracterización del desarrollo de proteínas mediante espectrometría de masas de reticulación rápida utilizando reticulantes di-orto-ftalaldehído. Comunicaciones de la naturaleza, 13 (1), 1468.