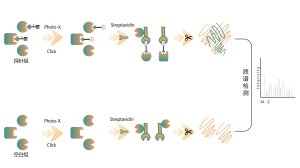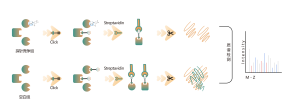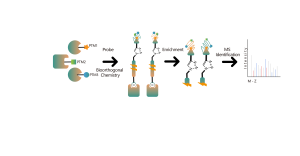
Productos
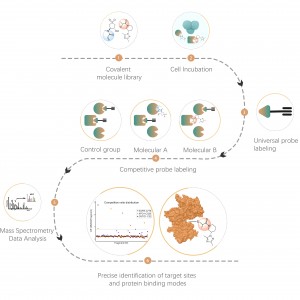
Identificación de objetivos de proteínas mediante proteómica diferencial
Características técnicas de la plataforma
La proteómica diferencial estudia los cambios del proteoma bajo diferentes estados fisiológicos o patológicos, como tratamientos farmacológicos o regulación genética, comparando dos o más muestras. Este enfoque arroja luz sobre procesos vitales importantes o enfermedades importantes para descubrir las diferentes proteínas clave que se consideran marcadores para el análisis cualitativo y funcional. Se pueden identificar cuantitativamente miles de proteínas con el protocolo estándar ChomiX, incluida la preparación de muestras de proteoma, digestión con proteasas, fraccionamiento de péptidos, adquisición de datos de EM y análisis bioinformático.

Oferta de servicios
1. Análisis cuantitativo y de todo el proteoma de proteínas diferenciales causadas por enfermedades, tratamientos farmacológicos o estrés ambiental, etc.
2. Análisis cuantitativo del proteoma de estructuras subcelulares (membranas celulares, núcleos, mitocondrias, etc.).
3. Descubrimiento de biomarcadores en toda la escala del proteoma.
Métodos de cuantificación proteómica
Cuantificación sin etiquetas (LFQ)
Técnica:
Cuantificación de proteínas por recuento espectral o intensidad XIC, cuantificación de nivel MS1
Ventajas:
sin etiquetado de isótopos, alto rendimiento
Requisitos de muestra:
Muestras de células, tejidos, sangre, etc.
Marcado de isótopos estables mediante dimetilación reductiva (ReDi)
Tecnología:
Se utilizan formas regulares (ligeras) o deuteradas (pesadas) de formaldehído y cianoborohidruro de sodio para agregar dos grupos metilo al extremo N del péptido y a la cadena lateral de los residuos de lisina. Cuantificación de nivel MS1
Ventajas:
etiquetado químico dúplex y triplex, bajo costo, velocidad de reacción rápida, alta reproducibilidad, sin limitación de muestras;
Requisitos de muestra:
Muestras de células, tejidos, sangre, etc.
Marcado de isótopos estables mediante aminoácidos en cultivo celular (SILAC)
Tecnología:
Cultivo celular en medio que contiene isótopos estables que marcan aminoácidos esenciales para la cuantificación del proteoma en diferentes muestras; Cuantificación de nivel MS1
Ventajas:
etiquetado metabólico dúplex, menos error del sistema;
Requisitos de muestra:
muestras de células vivas.
Etiquetas de masa en tándem (TMT/IBT)
Tecnología:
Cuantificación relativa de la intensidad del péptido por su grupo informador en diferentes muestras; Cuantificación a nivel MS2
Ventajas:
Cuantificación de hasta 16 muestras, cuantificación precisa;
Requisitos de muestra:
Muestras de células, tejidos, sangre, etc.
Leyenda de la figura
Objetivo del proyecto
Análisis comparativo de los cambios en los niveles de proteoma completo entre el grupo tratado con el fármaco y el grupo de control para investigar los mecanismos moleculares subyacentes al fenotipo del fármaco.
Tipos de muestra
Muestras celulares sometidas a tratamientos farmacológicos y de control, cada una compuesta por tres réplicas biológicas.
Método experimental
Identificación cuantitativa de proteínas expresadas diferencialmente a nivel de proteoma completo utilizando una metodología proteómica de etiquetado de isótopos múltiples basada en TMT.
Visualización de datos

Gráfico de volcán para análisis de abundancia de proteínas.
Como se muestra en el gráfico del volcán, se cuantificaron 5987 proteínas en un total de seis grupos. La proporción de cada proteína se analizó mediante análisis de prueba t que mostró 560 proteínas reguladas positivamente y 363 proteínas reguladas negativamente en el grupo tratado con el fármaco. La información de intensidad relacionada también se presentó en el mapa de calor.

- Mapa de calor de abundancia de proteínas entre el grupo control y experimental.
Se realizaron análisis de la vía KEGG y de ontología genética (GO) en las proteínas expresadas diferencialmente, incluido el proceso biológico GOTERM, el componente celular GOTERM y la función molecular GOTERM. Al evaluar el nivel de importancia del enriquecimiento de GOTERM, identificamos categorías funcionales y vías significativamente enriquecidas por las proteínas expresadas diferencialmente, contribuyendo así a la exploración de los mecanismos moleculares de los fármacos.

- Análisis de ontología genética de proteínas diferenciales.
El análisis de ontología genética, incluido GOTERM_ proceso biológico, GOTERM_ componente celular, GOTERM_ función molecular y vía KEGG, reveló que las proteínas reguladas positivamente se enriquecieron significativamente en la segregación de cromosomas nucleares, la segregación de cromátidas hermanas mitóticas y la vía de señalización de segregación de cromátidas hermanas.