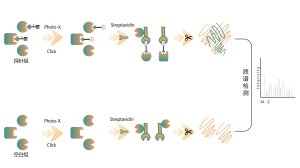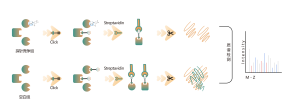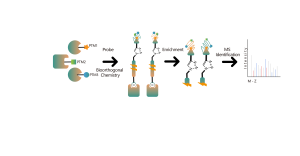
Produits
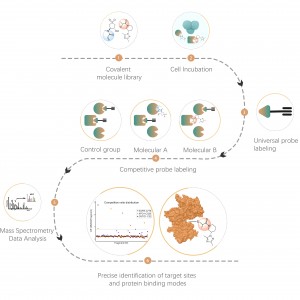
Identification des cibles protéiques par protéomique différentielle
Caractéristiques techniques de la plateforme
La protéomique différentielle étudie les modifications du protéome sous différents états physiologiques ou pathologiques, tels que des traitements médicamenteux ou la régulation génique, en comparant deux échantillons ou plus. Cette approche met en lumière des processus vitaux importants ou des maladies majeures afin de déterminer les différentes protéines clés considérées comme des marqueurs pour l'analyse qualitative et fonctionnelle. Des milliers de protéines peuvent être identifiées quantitativement avec le protocole standard ChomiX, notamment la préparation d'échantillons de protéome, la digestion par protéase, le fractionnement des peptides, l'acquisition de données MS et l'analyse bioinformatique.

Offre de services
1. Analyse quantitative et à l'échelle du protéome des protéines différentielles causées par des maladies, des traitements médicamenteux ou un stress environnemental, etc.
2. Analyse quantitative du protéome des structures subcellulaires (membranes cellulaires, noyaux, mitochondries, etc.).
3. Découverte de biomarqueurs à l'échelle complète du protéome.
Méthodes de quantification protéomique
Quantification sans étiquette (LFQ)
Technique:
Quantification des protéines par comptage spectral ou intensité XIC, quantification au niveau MS1
Avantages :
pas de marquage isotopique, débit élevé
Exemples d'exigences :
Cellules, tissus, échantillons de sang, etc.
Marquage des isotopes stables par diméthylation réductrice (ReDi)
Technologie:
Des formes régulières (légères) ou deutérées (lourdes) de formaldéhyde et de cyanoborohydrure de sodium sont utilisées pour ajouter deux groupes méthyle à l'extrémité N du peptide et à la chaîne latérale des résidus lysine. Quantification au niveau MS1
Avantages :
étiquetage chimique duplex et triplex, faible coût, taux de réaction rapide, reproductibilité élevée, aucune limitation des échantillons ;
Exemples d'exigences :
Cellules, tissus, échantillons de sang, etc.
Marquage des isotopes stables par acides aminés en culture cellulaire (SILAC)
Technologie:
Culture cellulaire dans un milieu contenant des isotopes stables marquant les acides aminés essentiels pour la quantification du protéome dans différents échantillons ; Quantification au niveau MS1
Avantages :
étiquetage métabolique duplex, moins d'erreurs système ;
Exemples d'exigences :
échantillons de cellules vivantes.
Balises de masse tandem (TMT/IBT)
Technologie:
Quantification relative de l'intensité du peptide par son groupe rapporteur dans différents échantillons ; Quantification au niveau MS2
Avantages :
Quantification jusqu'à 16 échantillons, quantification précise ;
Exemples d'exigences :
Cellules, tissus, échantillons de sang, etc.
Légende des figures
Objectif du projet
Analyse comparative des changements dans les niveaux de protéome entier entre le groupe traité par le médicament et le groupe témoin pour étudier les mécanismes moléculaires sous-jacents au phénotype du médicament.
Types d'échantillons
Échantillons cellulaires soumis à des traitements médicamenteux et de contrôle, chacun comprenant trois répétitions biologiques.
Méthode expérimentale
Identification quantitative de protéines différentiellement exprimées au niveau du protéome entier à l'aide de la méthodologie protéomique de marquage isotopique multiple basée sur le TMT.
Visualisation des données

Graphique volcanique pour l'analyse de l'abondance des protéines
Comme le montre le tracé du volcan, 5 987 protéines ont été quantifiées dans un total de six groupes. Le rapport de chaque protéine a été analysé par analyse de test t montrant 560 protéines régulées positivement et 363 protéines régulées négativement dans le groupe traité par le médicament. Les informations d’intensité associées ont également été présentées dans la carte thermique.

- Carte thermique de l'abondance des protéines entre le groupe témoin et le groupe expérimental.
Des analyses de la voie KEGG et de l'ontologie génétique (GO) ont été effectuées sur les protéines exprimées différentiellement, notamment GOTERM-Biological Process, GOTERM-Cellular Component et GOTERM-Molecular Function. En évaluant le niveau significatif de l'enrichissement de GOTERM, nous avons identifié des catégories fonctionnelles et des voies significativement enrichies par les protéines différentiellement exprimées, contribuant ainsi à l'exploration des mécanismes moléculaires des médicaments.

- Analyse de l'ontologie génétique des protéines différentielles.
L'analyse de l'ontologie génétique, y compris le processus biologique GOTERM_, le composant cellulaire GOTERM_, la fonction moléculaire GOTERM_ et la voie KEGG, a révélé que les protéines régulées positivement étaient enrichies de manière significative dans la ségrégation des chromosomes nucléaires, la ségrégation des chromatides sœurs mitotiques et la voie de signalisation de la ségrégation des chromatides sœurs.