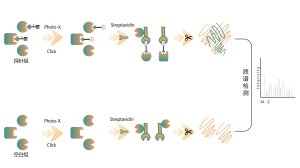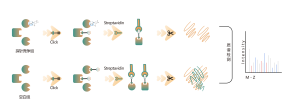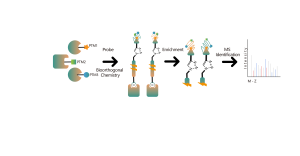
Prodotti
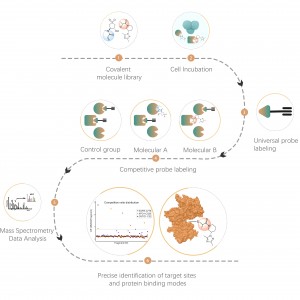
Identificazione di bersagli proteici mediante proteomica differenziale
Caratteristiche tecniche della piattaforma
La proteomica differenziale studia i cambiamenti del proteoma in diversi stati fisiologici o patologici, come trattamenti farmacologici o regolazione genetica, confrontando due o più campioni. Questo approccio fa luce su importanti processi vitali o sulle principali malattie al fine di individuare le diverse proteine chiave considerate marcatori per l'analisi qualitativa e funzionale. Migliaia di proteine possono essere identificate quantitativamente con il protocollo standard ChomiX che comprende la preparazione del campione del proteoma, la digestione delle proteasi, il frazionamento dei peptidi, l'acquisizione di dati MS e l'analisi bioinformatica.

Offerta di servizi
1. Analisi quantitativa e proteome-wide di proteine differenziali causate da malattie, trattamenti farmacologici o stress ambientale ecc..
2. Analisi quantitativa del proteoma di strutture subcellulari (membrane cellulari, nuclei, mitocondri, ecc.).
3. Scoperta di biomarcatori nell'intera scala del proteoma.
Metodi di quantificazione proteomica
Quantificazione senza etichetta (LFQ)
Tecnica:
Quantificazione delle proteine mediante conteggio spettrale o intensità XIC, quantificazione a livello MS1
Vantaggi:
nessuna etichettatura isotopica, elevata produttività
Requisiti del campione:
Campioni di cellule, tessuti, sangue, ecc.
Etichettatura degli isotopi stabili mediante dimetilazione riduttiva (ReDi)
Tecnologia:
Le forme regolari (leggere) o deuterate (pesanti) di formaldeide e cianoboroidruro di sodio vengono utilizzate per aggiungere due gruppi metilici all'estremità N terminale del peptide e alla catena laterale dei residui di lisina. Quantificazione a livello MS1
Vantaggi:
etichettatura chimica duplex e triplex, basso costo, velocità di reazione rapida, elevata riproducibilità, nessuna limitazione dei campioni;
Requisiti del campione:
Campioni di cellule, tessuti, sangue, ecc.
Etichettatura degli isotopi stabili mediante aminoacidi in colture cellulari (SILAC)
Tecnologia:
Coltura cellulare in terreno contenente isotopi stabili che marcano gli aminoacidi essenziali per la quantificazione del proteoma in diversi campioni; Quantificazione a livello MS1
Vantaggi:
etichettatura metabolica duplex, meno errori di sistema;
Requisiti del campione:
campioni di cellule viventi.
Tag di massa tandem (TMT/IBT)
Tecnologia:
Quantificazione relativa dell'intensità del peptide da parte del suo gruppo reporter in diversi campioni; Quantificazione a livello MS2
Vantaggi:
Fino a 16 quantificazione dei campioni, quantificazione accurata;
Requisiti del campione:
Campioni di cellule, tessuti, sangue, ecc.
Legenda della figura
Scopo del progetto
Analisi comparativa dei cambiamenti nei livelli dell'intero proteoma tra il gruppo trattato con il farmaco e il gruppo di controllo per indagare i meccanismi molecolari alla base del fenotipo del farmaco.
Tipi di campioni
Campioni cellulari sottoposti a trattamenti farmacologici e di controllo, ciascuno comprendente tre repliche biologiche.
Metodo sperimentale
Identificazione quantitativa delle proteine espresse in modo differenziale a livello dell'intero proteoma utilizzando la metodologia proteomica di etichettatura di isotopi multipli basata su TMT.
Visualizzazione dei dati

Grafico del vulcano per l'analisi dell'abbondanza di proteine
Come mostrato nel grafico del vulcano, sono state quantificate 5987 proteine in un totale di sei gruppi. Il rapporto di ciascuna proteina è stato analizzato mediante analisi t-test che ha mostrato 560 proteine sovraregolate e 363 proteine sottoregolate nel gruppo trattato con il farmaco. Le relative informazioni sull'intensità sono state presentate anche nella mappa termica.

- Mappa termica dell'abbondanza proteica tra controllo e gruppo sperimentale.
Sono state condotte analisi del percorso KEGG e di Gene Ontology (GO) sulle proteine espresse in modo differenziale, tra cui GOTERM-Processo biologico, GOTERM-Componente cellulare e GOTERM-Funzione molecolare. Valutando il livello di significatività dell'arricchimento GOTERM, abbiamo identificato categorie funzionali e percorsi significativamente arricchiti dalle proteine espresse in modo differenziale, contribuendo così all'esplorazione dei meccanismi molecolari dei farmaci.

- Analisi di ontologia genetica delle proteine differenziali.
L'analisi dell'ontologia genetica, inclusa GOTERM_ Processo biologico, GOTERM_ Componente cellulare, GOTERM_ Funzione molecolare e percorso KEGG, ha rivelato che le proteine sovraregolate erano significativamente arricchite nella segregazione dei cromosomi nucleari, nella segregazione dei cromatidi fratelli mitotici e nella via di segnalazione della segregazione dei cromatidi fratelli.