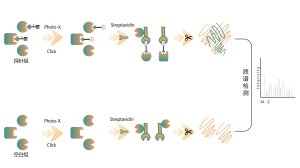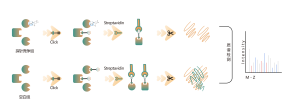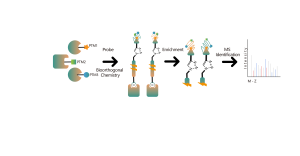
제품
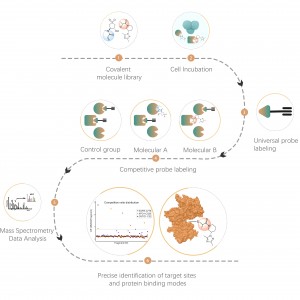
차등 단백질체학을 통한 단백질 표적 식별
플랫폼 기술적 특징
Differential proteomics는 두 개 이상의 샘플을 비교하여 약물 치료나 유전자 조절과 같은 다양한 생리학적 또는 병리학적 상태에서 프로테옴의 변화를 연구합니다. 이 접근법은 질적 및 기능적 분석을 위한 지표로 간주되는 주요 단백질을 파악하기 위해 중요한 생활 과정이나 주요 질병에 대해 조명합니다. 프로테옴 샘플 준비, 프로테아제 분해, 펩타이드 분획, MS 데이터 수집 및 생물정보학 분석을 포함한 ChomiX 표준 프로토콜을 사용하여 수천 개의 단백질을 정량적으로 식별할 수 있습니다.

서비스 제공
1. 질병, 약물 치료 또는 환경 스트레스 등에 의해 발생하는 차별적인 단백질의 정량적, 단백체 전반에 걸친 분석.
2. 세포내 구조(세포막, 핵, 미토콘드리아 등)의 단백질체를 정량 분석합니다.
3. 전체 프로테옴 규모의 바이오마커 발굴.
단백질 정량화 방법
LFQ(무표지 정량)
기술:
스펙트럼 카운트 또는 XIC 강도에 의한 단백질 정량화, MS1 수준 정량화
장점:
동위원소 라벨링 없음, 높은 처리량
샘플 요구사항:
세포, 조직, 혈액 샘플 등
환원적 디메틸화(ReDi)를 통한 안정 동위원소 표지
기술:
일반(경질) 또는 중수소화(중) 형태의 포름알데히드와 시아노수소화붕소나트륨은 펩타이드 N 말단과 리신 잔기의 측쇄에 2개의 메틸기를 추가하는 데 사용됩니다. MS1 수준 정량화
장점:
이중 및 삼중 화학 라벨링, 저렴한 비용, 빠른 반응 속도, 높은 재현성, 샘플 제한 없음;
샘플 요구사항:
세포, 조직, 혈액 샘플 등
세포 배양 내 아미노산을 이용한 안정 동위원소 표지(SILAC)
기술:
다양한 샘플에서 프로테옴 정량화를 위해 필수 아미노산을 표시하는 안정 동위원소를 함유한 배지에서 세포 배양 MS1 수준 정량화
장점:
이중 대사 라벨링, 시스템 오류 감소;
샘플 요구사항:
살아있는 세포 샘플.
탠덤 질량 태그(TMT/IBT)
기술:
다양한 샘플에서 리포터 그룹에 의한 펩타이드 강도의 상대적 정량화 MS2 수준 정량화
장점:
최대 16개의 샘플 정량화, 정확한 정량화;
샘플 요구사항:
세포, 조직, 혈액 샘플 등
그림 범례
프로젝트 목표
약물 표현형의 기본 분자 메커니즘을 조사하기 위해 약물 처리 그룹과 대조군 간의 전체 프로테옴 수준의 변화를 비교 분석합니다.
샘플 유형
약물 및 대조 처리를 거친 세포 표본은 각각 3개의 생물학적 복제물로 구성됩니다.
실험방법
TMT 기반 다중 동위원소 라벨링 프로테오믹스 방법론을 사용하여 전체 프로테옴 수준에서 차별적으로 발현된 단백질을 정량적으로 식별합니다.
데이터 시각화

단백질 풍부도 분석을 위한 화산 플롯
화산 플롯에서 볼 수 있듯이 총 6개 그룹에서 5987개의 단백질이 정량화되었습니다. t-test 분석을 통해 각 단백질의 비율을 분석한 결과 약물 투여군에서 상향조절된 단백질은 560개, 하향조절된 단백질은 363개였다. 관련 강도 정보도 히트맵에 표시되었습니다.

- 대조군과 실험군 사이의 단백질 풍부도에 대한 히트맵.
GOTERM-생물학적 과정, GOTERM-세포 구성 요소 및 GOTERM-분자 기능을 포함하여 차등적으로 발현된 단백질에 대해 KEGG 경로 및 유전자 존재론(GO) 분석이 수행되었습니다. GOTERM 농축의 중요성 수준을 평가함으로써 우리는 차등적으로 발현된 단백질에 의해 상당히 강화된 기능적 범주와 경로를 식별하여 약물 분자 메커니즘의 탐색에 기여했습니다.

- 차등 단백질의 유전자 온톨로지 분석.
GOTERM_ 생물학적 과정, GOTERM_ 세포 구성 요소, GOTERM_ 분자 기능 및 KEGG 경로를 포함한 유전자 온톨로지 분석을 통해 상향 조절된 단백질이 핵 염색체 분리, 유사분열 자매 염색분체 분리 및 자매 염색분체 분리 신호 전달 경로에서 상당히 풍부하다는 사실이 밝혀졌습니다.