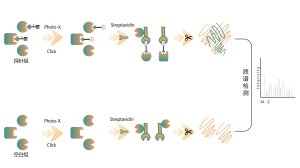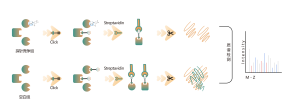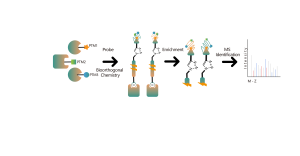
Produkty
Identyfikacja celów białkowych metodą proteomiki różnicowej
Funkcje techniczne platformy
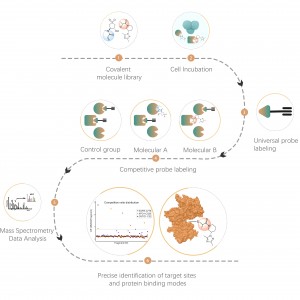
Proteomika różnicowa bada zmiany proteomu w różnych stanach fizjologicznych lub patologicznych, takich jak leczenie farmakologiczne lub regulacja genów, poprzez porównanie dwóch lub więcej próbek. Podejście to rzuca światło na ważne procesy życiowe lub główne choroby, umożliwiając określenie kluczowych różnych białek, które są uważane za markery analizy jakościowej i funkcjonalnej. Tysiące białek można zidentyfikować ilościowo za pomocą standardowego protokołu ChomiX, obejmującego przygotowanie próbki proteomu, trawienie proteazą, frakcjonowanie peptydów, gromadzenie danych MS i analizę bioinformatyczną.

Oferta usług
1. Ilościowa i obejmująca cały proteom analiza zróżnicowanych białek spowodowanych chorobami, leczeniem farmakologicznym, stresem środowiskowym itp.
2. Analiza ilościowa proteomu ze struktur subkomórkowych (błony komórkowe, jądra, mitochondria itp.).
3. Odkrycie biomarkerów w skali całego proteomu.
Metody kwantyfikacji proteomicznej
Oznaczanie ilościowe bez etykiet (LFQ)
Technika:
Kwantyfikacja białka poprzez zliczenie widma lub intensywność XIC, kwantyfikacja na poziomie MS1
Zalety:
brak znakowania izotopowego, wysoka przepustowość
Przykładowe wymagania:
Komórki, tkanki, próbki krwi itp.
Znakowanie stabilnych izotopów metodą redukcyjnej dimetylacji (ReDi)
Technologia:
W celu dodania dwóch grup metylowych do N-końca peptydu i łańcucha bocznego reszt lizyny stosuje się zwykłe (lekkie) lub deuterowane (ciężkie) formy formaldehydu i cyjanoborowodorku sodu. Kwantyfikacja na poziomie MS1
Zalety:
znakowanie chemiczne metodą duplex i triplex, niski koszt, szybka reakcja, wysoka powtarzalność, brak ograniczeń dotyczących próbek;
Przykładowe wymagania:
Komórki, tkanki, próbki krwi itp.
Stabilne znakowanie izotopów przez aminokwasy w hodowli komórkowej (SILAC)
Technologia:
Hodowla komórkowa w pożywce zawierającej stabilny izotop znakujący niezbędne aminokwasy do ilościowego oznaczania proteomu w różnych próbkach; Kwantyfikacja na poziomie MS1
Zalety:
dupleksowe znakowanie metaboliczne, mniej błędów systemowych;
Przykładowe wymagania:
próbki żywych komórek.
Tandemowe znaczniki masowe (TMT/IBT)
Technologia:
Względne oznaczenie ilościowe intensywności peptydu przez jego grupę reporterową w różnych próbkach; Kwantyfikacja na poziomie MS2
Zalety:
Kwantyfikacja do 16 próbek, dokładna ocena ilościowa;
Przykładowe wymagania:
Komórki, tkanki, próbki krwi itp.
Legenda rysunku
Cel projektu
Analiza porównawcza zmian w poziomach całego proteomu pomiędzy grupą leczoną lekiem a grupą kontrolną w celu zbadania mechanizmów molekularnych leżących u podstaw fenotypu leku.
Typy próbek
Próbki komórkowe poddane działaniu leku i terapii kontrolnej, każda zawierająca trzy repliki biologiczne.
Metoda eksperymentalna
Ilościowa identyfikacja białek ulegających różnej ekspresji na poziomie całego proteomu przy użyciu metodologii proteomiki wieloizotopowego znakowania opartej na TMT.
Wizualizacja danych

Wykres wulkanu do analizy obfitości białka
Jak pokazano na wykresie wulkanu, w sumie oznaczono ilościowo 5987 białek w sześciu grupach. Stosunek każdego białka analizowano za pomocą analizy testu t wykazującej podwyższoną ekspresję 560 białek i 363 białek obniżoną w grupie leczonej lekiem. Powiązane informacje o intensywności zostały również przedstawione na mapie cieplnej.

- Mapa cieplna obfitości białka pomiędzy grupą kontrolną i eksperymentalną.
Przeprowadzono analizy szlaku KEGG i ontologii genów (GO) na białkach o różnej ekspresji, w tym na procesie biologicznym GOTERM, składniku komórkowym GOTERM i funkcji molekularnej GOTERM. Oceniając poziom istotności wzbogacenia GOTERM, zidentyfikowaliśmy kategorie funkcjonalne i ścieżki znacząco wzbogacone przez białka o różnej ekspresji, przyczyniając się w ten sposób do badania mechanizmów molekularnych leków.

- Analiza genów Ontologia białek różnicowych.
Analiza genów, w tym proces biologiczny GOTERM_, składnik komórkowy GOTERM_, funkcja molekularna GOTERM_ i szlak KEGG, ujawniła, że białka o zwiększonej ekspresji były znacząco wzbogacone w segregację chromosomów jądrowych, mitotyczną segregację chromatyd siostrzanych i szlak sygnalizacyjny segregacji chromatyd siostrzanych.