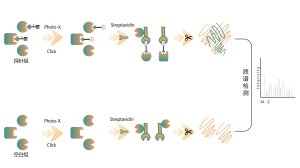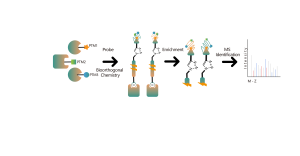Products
Protein Targets Identification by Differential Proteomics
Platform Technical Features
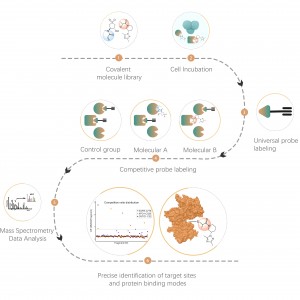
Differential proteomics studies the changes of proteome under different physiological or pathological states, such as drug treatments or gene regulation, by comparing two or more samples. This approach sheds light on important life processes or major diseases in order to figure out the key different proteins that are regarded as markers for qualitative and functional analysis. Thousands of proteins can be identified quantitatively with the ChomiX standard protocol including proteome sample preparation, protease digestion, peptide fractionation, MS data acquisition and bioinformatics analysis.

Service Offering
1. Quantitative and proteome-wide analysis of differential proteins caused by diseases, drug treatments or environmental stress etc..
2. Quantitative analysis of proteome from subcellular structures (cell membranes, nuclei, mitochondria, etc.).
3. Discovery of biomarkers in the whole proteome scale.
Proteomic Quantification Methods
Label-Free Quantitation (LFQ)
Technique:
Protein quantification by spectral count or XIC intensity, MS1-level quantification
Advantages:
no isotope labeling, high throughput
Sample requirements:
Cell, tissue, blood samples etc.
Stable Isotope Labeling by Reductive Dimethylation (ReDi)
Technology:
Either regular (light) or deuterated (heavy) forms of formaldehyde and sodium cyanoborohydride are used to add two methyl groups to the peptide N terminus and the side chain of lysine residues. MS1-level quantification
Advantages:
duplex and triplex chemical labelling, low cost, fast reaction rate, high reproducibility, no limitation of samples;
Sample requirements:
Cell, tissue, blood samples etc.
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
Technology:
Cell culture in medium containing stable isotope labeling essential amino acids for proteome quantification in different samples; MS1-level quantification
Advantages:
duplex metabolic labeling, less system error;
Sample requirements:
living cell samples.
Tandem Mass Tags (TMT/IBT)
Technology:
Relative quantification of peptide intensity by its reporter group in different samples; MS2-level quantification
Advantages:
Up to 16 samples quantification, accurate quantification;
Sample requirements:
Cell, tissue, blood samples etc.
Figure Legend
Project Aim
Comparative analysis of changes in whole proteome levels between the drug-treated group and control group to investigate molecular mechanisms underlying drug phenotype.
Sample Types
Cellular specimens subjected to drug and control treatments, each comprising three biological replicates.
Experimental Method
Quantitative identification of differentially expressed proteins at the whole proteome level using TMT-based multiple isotope labeling proteomics methodology.
Data Visualization

Volcano plot for protein abundance analysis
As shown in the volcano plot, 5987 proteins were quantified in total six groups. The ratio of each protein was analyzed by t-test analysis showing 560 proteins upregulated and 363 proteins downregulated in the drug-treated group. The related intensity information was also presented in the heatmap.

- Heatmap of protein abundance between control and experimental group.
KEGG pathway and Gene Ontology (GO) analyses were conducted on the differentially expressed proteins, including GOTERM-Biological Process, GOTERM-Cellular Component, and GOTERM-Molecular Function. By assessing the significance level of GOTERM enrichment, we identified functional categories and pathways significantly enriched by the differentially expressed proteins, thereby contributing to the exploration of drug molecular mechanisms.

- Gene Ontology analysis of differential proteins.
Gene Ontology analysis, including GOTERM_ Biological process, GOTERM_ Cellular component, GOTERM_ Molecular function and KEGG pathway, revealed that the upregulated proteins were significantly enriched in nuclear chromosome segregation, mitotic sister chromatid segregation, and sister chromatid segregation signaling pathway.